PRESENTACIÓN DE CASO
Sarcoma pleomórfico. Presentación de un caso
Pleomorphic sarcoma. Case presentation
Dr. Enrique A. Pancorbo Sandoval,I Dr. Alberto Delgado Quiñones,I Dr. Giraldo Díaz Prieto,I Dr. Justo Hernández Hernández,I Dr. Luis A. Pinto ContrerasII
I Hospital Militar Docente Dr. Mario Muñoz Monroy. Matanzas, Cuba.
II Hospital Provincial Docente Clínico Quirúrgico José Ramón López Tabrane. Servicios de Oncología. Matanzas, Cuba.
RESUMEN
Se presenta un paciente del sexo masculino de 36 años de edad, que en el momento del inicio del cuadro clínico, a partir de un trauma leve, debutó con fiebre, dolor y aumento de temperatura a nivel del muslo derecho, se valoró como un posible hematoma abscedado por lo que requirió ingreso y tratamiento quirúrgico. Posterior al alta y dos meses de evolución, regresa al centro hospitalario por presentar una evolución tórpida con un aumento de volumen de tipo tumoral se ingresó, estudió y se valoró por un equipo de especialistas multidisciplinarios decidiendo una nueva intervención quirúgica en base a los resultados de laboratorio y estudios imagino lógicos, se le realizó exéresis de la tumoración (a nivel del vasto externo) con margen oncológico y enviado para anatomía patológica, informado como un sarcoma pleomórfico, llevó posteriormente tratamiento adyuvante de quimioterapia, contando con 10 años de sobrevivencia en la actualidad y totalmente asintomático.
Palabras clave: sarcoma pleomórfico muslo derecho, exéresis de la tumoración, tratamiento multidisciplinario.
ABSTRACT
The case of a male patient aged 36 years is presented. The clinical characteristics are the following: after a light trauma, he had fever, pain, temperature increase in the right thigh. It was diagnosed as a possible abscessed hematoma and the patient was admitted in the hospital and surgically treated. After discharging and two-months evolution, he came back to the hospital presenting a torpid evolution with a tumoral-kind volume increase. He was readmitted, studied and evaluated by a team of multidiscipline specialists, who decided a new surgical intervention based on the laboratory and imaging studies. Tumor was removed (at the level of external vastus) with oncologic margin and send to pathologic anatomy. It was informed as a pleomorphic sarcoma. An adjuvant chemotherapeutic treatment was applied later. Currently, the patient has survived 10 years and is totally asymptomatic.
Key words: pleomorphic sarcoma, right thigh, tumor removal, multidiscipline treatment.
INTRODUCCIÓN
Los sarcomas de partes blandas (SPB) constituyen un grupo amplio y heterogéneo de tumores poco comunes, que se caracterizan por requerir un tratamiento multidisciplinario frecuentemente complejo. En los últimos años, se han producido avances notables en el conocimiento de la patología y la biología molecular de esta enfermedad, el tratamiento clínico ha evolucionado de forma más discreta y, en la práctica, debido principalmente a la rareza y complejidad de esta enfermedad, los resultados no son siempre los óptimos.
Los sarcomas son un grupo amplio de tumores malignos que pueden aparecer casi en cualquier parte del organismo y afectan a personas jóvenes y ancianas por igual. Los sarcomas de tejidos blandos pueden originarse en todo tipo de tejidos blandos del organismo, incluidos nervios, grasa, músculo y vasos sanguíneos. Los sarcomas también pueden aparecer, prácticamente cualquier órgano, como pulmones, corazón, tubo digestivo, hígado, riñones y extremidades.
Más de la mitad de los sarcomas de tejidos blandos, alrededor del 60 %, aparecen en los brazos y las piernas. Otros focos habituales de sarcoma son el tronco (20 % de los casos), el abdomen (15 %) y la región de la cabeza y el cuello.(1)
PRESENTACIÓN DEL CASO
Paciente de 36 años de edad, sexo masculino, que acude al cuerpo de guardia del Hospital Militar de Matanzas, por primera vez, por dolor y aumento de volumen con calor local en la cara externa del muslo derecho, con el antecedente, referido por el paciente, de un trauma leve a nivel de la cara externa del muslo derecho distal al trocánter mayor de la cadera; siendo interpretado por el ortopédico de guardia como un hematoma abscedado ingresándolo y llevándolo al salón de operaciones para drenar el mismo, evacuando solamente un líquido serohemático, observando un cambio de coloración grisáceo del músculo vasto externo como carne de pescado, estando ingresado durante 7 días con tratamiento de antibiótico fue dado alta hospitalaria con seguimiento por consulta.
Siendo ingresado por segunda vez a los 2 meses ya que el cuadro clínico doloroso continuaba, acompañado de fiebre, taquipnea, anorexia y pérdida de peso. Al examen físico se palpó una tumoración mayor de 7 cm, adherido al plano profundo, a nivel de la cicatriz anterior en el muslo derecho cara externa, aumento de la temperatura local, con ganglios aumentados de tamaño en región inguinal derecha. Al examen físico abdominal todo fue negativo.
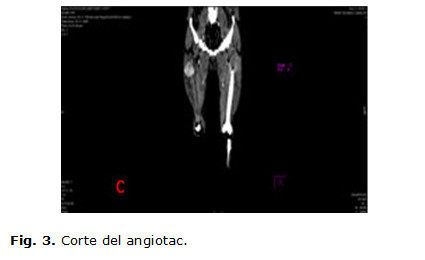
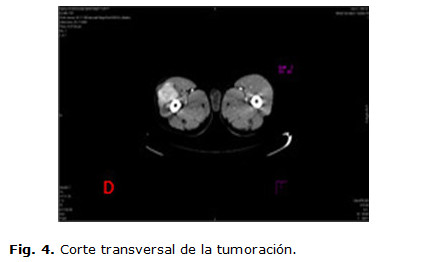
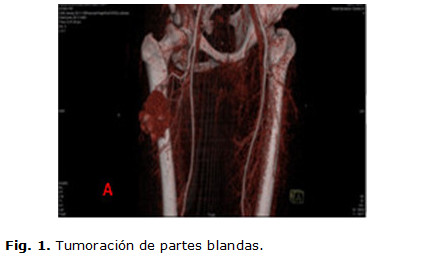
Se realizan estudios hematológicos y hemoquímicos donde se informan: hemoblobina en 11 g/l, eritrosidimentación en 122 mm/h, lámina periférica con plaquetas adecuadas, el leucograma dentro de cifras normales con neutrófilos elevados, radiografía de tórax con refuerzo de la trama vascular, radiografía de senos peri nasales con imagen de aspecto polipoideo en seno maxilar derecho, el ultrasonido de partes blanda informó posible absceso de 7 cm con imagen hipoecogénica central, fue efectuado una biopsia por aspiración siendo no útil, por lo cual se decide indicar una tomografía computada (TC) de 99 cortes contrastada, informándose una colección muy celular que interesa el vasto externo midiendo más o menos 7 cm muy vascularizado (figuras 1,2,3,4), con todos estos resultados se discute en colectivo con el servicio de oncología y el radiólogo que efectuó el angiotac, descartando la posibilidad de efectuar una embolización del tumor ya que la tumoración se nutría fundamentalmente, de la arteria femoral profunda, ya que no presentaba lesiones metastásicas en los diferentes estudios efectuados, se decide realizar una exéresis de la tumoración con margen oncológico para biopsia y toma de ganglios de la región inguinal proximal; y en un segundo tiempo inmediato tratamiento adyuvante con quimioterapia por el servicio de oncología. Posterior al acto quirúrgico el cuadro febril y respiratorio remitió espontáneamente, mejorando el estado general del paciente.
El tejido tomado para biopsia que incluyó gran parte del músculo vasto externo mostraba macroscópicamente en el interior del mismo una masa grisácea (como carne de pescado) que por su aspecto celular indiferenciado fue informado como un sarcoma pleomórfico por el servicio de anatomía patológica con posibilidades diagnósticas de un liposarcoma pleomórfico, en primer lugar y de un fibrohistiocitoma en segundo lugar, siendo informado los ganglios tomados como una adenitis inespecífica. Por lo cual, según la Clasificación TNM1 para sarcoma de partes blandas fue informado como un T2b, N0, M0 de grado alto, grado 4 y estadío III.
El paciente durante estos 10 años ha tenido seguimiento especializado sin recidivas de ningún proceso anárquico proliferativo.
DISCUSIÓN
Hay más de 30 tipos diferentes de sarcomas de tejidos blandos malignos. Pueden clasificarse simplemente según los tipos de células afectadas: grasa, músculo liso, músculo esquelético, tejido nervioso, articulaciones, vasos sanguíneos o linfáticos y tejido fibroso. También hay una categoría de sarcomas de tipo indeterminado o mixto.
El sarcoma pleomorfo y el histiocitoma fibroso maligno (HFM) aparecen en el tejido fibroso de los brazos y las piernas. Es una forma frecuente de sarcoma maligno en ancianos y tiende a diseminarse a zonas distantes del cuerpo, sobre todo los pulmones2. Aunque es visto igualmente en pacientes jóvenes.(1)
A pesar de ser una afección que no aparece con una frecuencia alarmante, es una preocupación para todo cirujano enfrentar dicha entidad. Lo cual ha provocado en los últimos años, que en diferentes partes del mundo se oriente a crear centros de referencias con personal multidisciplinario para dar mejor respuesta a dicha tumoración y lograr una mayor supervivencia de los enfermos que sean portadores de un sarcoma.(1-5)
Los sarcomas de tejido blando son tumores malignos que surgen en cualquiera de los tejidos mesodérmicos de las extremidades (50 %), el tronco y el retroperitoneo (40 %), o de la cabeza y el cuello (10 %). Se cuenta con informes de que la tasa de incidencia internacional oscila de 1,8 a 5 por cada 100.000 por año.(1,3)
Los sarcomas de tejido blando se presentan con mayor frecuencia en pacientes con los siguientes síndromes hereditarios:(4-6)
Es importante que los cirujanos ortopédicos estén alertas frente a este tipo de sarcoma, y desde el punto de vista clínico considerar determinados signos de alarma, los cuales fueron propuestos por el Grupo Español de Investigación de Sarcomas (GEIS), constituido por un grupo multidisciplinario de expertos en el estudio y el tratamiento de los sarcomas, como una herramienta para orientarse con pacientes que acudan a los servicios de ortopedia.(1)
Deberán considerarse potencialmente malignas las tumoraciones que afecten partes blandas con cualquiera de los signos siguientes:
1. Tumoraciones mayores de 5 cm.
2. Tumoraciones que hayan experimentado un crecimiento reciente.
3. Tumoraciones profundas (fijas).
El dolor no es un síntoma que por sí oriente pero junto con cualquiera de los signos anteriores puede reforzar la presunción de malignidad.
Hay protocolos,(1,7) para el tratamiento quirúrgico de los sarcomas de tejidos blandos proponen que los márgenes deben ser de 5 cm, aunque se acepta hasta de 1 cm si la fascia que recubre el tumor está sana, el cual debe ser valorado previamente por un grupo multidisciplinario con vista a la planificación del proceder a seguir,(1,2, 7-9) donde en base a la experiencia de los últimos años generalmente, se planifica primero el tratamiento quirúrgico seguido de la combinación o no de radioterapia (lo que ha demostrado en los sarcomas pleomórficos no tener buenos resultados), pero sí la combinación de quimioterapia adyuvante ha demostrado ser más efectiva.(1-3,6,7,8)
La realización de un régimen de seguimiento consistente en un estudio semestral durante el primer año y anual hasta completar 5 años, de RM de la región tumoral, ha demostrado ser coste-efectivo, al menos, en los casos con tumores de grado alto o de grado bajo con márgenes de resección afectados.
En estudios efectuados donde se aplicó quimioterapia que incluía epirrubina e ifosfamida a dosis altas en SPB localizados en extremidades, cintura escapular o pélvica de grado alto, tamaño superior a 5 cm y localización profunda tras una media de seguimiento de 5 años, la supervivencia global fue significativamente superior. Sin embargo en un análisis posterior con seguimiento de 7,5 años, las diferencias en supervivencia dejaron de ser significativas.(9) Por último, un nuevo metanálisis actualizado que incluye a 18 ensayos aleatorizados ha mostrado un efecto significativo, aunque marginal en un (6 %), de la quimioterapia sobre la supervivencia global lo que indica que la asociación de doxorrubicina e ifosfamida puede tener un impacto mayor en la supervivencia que doxorrubicina sola.(10)
Los factores pronósticos más importantes para los sarcomas de partes blandas son: tamaño tumoral, grado de diferenciación e invasión ganglionar o metastásica.(11)
El seguimiento de los sarcomas (SPB) en extremidades no está bien definido en la actualidad, ya que el criterio de valoración radiográfica como en los sarcomas de tórax(1) no se comporta de igual manera. La resonancia magnética (RM) con contraste intravenoso de la región de la lesión primaria es la técnica de elección, si hay sospecha clínica de recidiva local. En caso de contraindicación de la RM, debería realizarse una TAC con contraste intravenoso.
En la visita de los 2-3 meses se recomienda obtener una RM del lugar de la lesión para disponer de un mapa posquirúrgico. Es importante que la RM se realice unas 8-10 semanas después de la intervención quirúrgica, para minimizar los falsos positivos debidos a cambios edematosos y hemorrágicos.
Algunos autores abogan por la realización anual de una RM durante los primeros 5 años de seguimiento posquirúrgico para detectar recidivas locales sólo en los casos de tumores de grado alto.
REFERENCIAS BIBLIOGRÁFICAS
1- García del Muro X, Martín J, Maurel J, Cubedo R, Bagué S, De Alavá E, et al. Guía de prácticas clínicas en los sarcomas de partes blandas. Med Clin [Internet]. 2011 [citado 12 Ene 2015];136(9):408.e1-408.e18. Disponible en: http://www.sciencedirect.com/science/article/pii/S002577531100176X
2- American Cancer Society [Internet]. Atlantac, Ga: American Cancer Society; c2015 [citado 12 Ene 2015]. Cancer Facts and Figures 2012. Disponible en: http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-figures
3- Wibmer C, Leithner A, Zielonke N, Sperl M, Windhager R. Increasing incidence rates of soft tissue sarcomas? A population-based epidemiologic study and literature review. Ann Oncol. 2010;21(5): 1106-11. Citado en PubMed; PMID: 19858086.
4- Singer S, Nielsen T, Antonescu CR. Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA. Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2010. p. 1522-32.
5- Singer S, Maki RG, O'Sullivan B. Soft tissue sarcoma. In: De Vita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2010. p. 1533-77.
6- Malawer MM, Helman LJ, O'Sullivan B. Sarcomas of bone. In: De Vita VT Jr, Lawrence TS, Rosenberg SA. Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2010. p. 1578-1609.
7- Valdés C, Oleada L, López I, Puertas J, Egilior J, Ortiz J, et al. Protocolo para diagnóstico y tratamiento de sarcomas de tejidos blandos del Hospital de Basurto. Cirugía Plástica Ibero-latinoamericana. 2004;30(4):285-92. Disponible en: http://filacp.org/web/espanol/revista-pdf/novenaentrega/valdes.pdf
8- Collazo Álvarez H, Torrecilla Silverio D, Morales Florat JL, Collazo Marín SY. Histiocitoma fibroso maligno. Presentación de caso. Rev Cubana de Ortop y Traumatol [Internet]. 2012 [citado 12 Ene 2015];26(1) 64-75. Disponible en: http://scielo.sld.cu/scielo.php?pid=S0864-215X2012000100007&script=sci_arttext&tlng=en
9- Frustaci S, De Paoli A, Bidoli E, La Mura N, Berretta M, Buonadonna A, et al. Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology. 2003;65(Suppl 2):80–4. Citado en PubMed; PMID: 14586155.
10- Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. Asystematic meta-analysis of randomized controlled trials of adjuvant chemotherapyfor localized resectable soft-tissue sarcoma. Cancer. 2008;113:573–81.Citado en PubMed; PMID:18521899.
11- Fletcher CD. World Health Organization Classification of Tumors. En: Pathology and Genetics. Tumors of Soft Tissue and Bone. Lyon: IARC Press; 2002.
Recibido: 10 de abril del 2015.
Aceptado: 15 de julio del 2015.
Dr. Enrique A. Pancorbo Sandoval. Hospital Militar Docente Dr. Mario Muñoz Monroy. Carretera Central. km 109 Gelpy. Reparto 2 de diciembre. Matanzas, Cuba. Correo electrónico: enriquepancorbo.mtz@infomed.sld